
Unique device identification (UDI) 是美国食品药物管理局FDA建立的”特殊医疗器械的识别系统”,该注册码的实施是为了有效识别在美国市场上销售并使用的医疗器械,无论其生产地在哪里。一旦实施,NHRIC和NDC标签将废止,所有的医疗器械都需要将这个新的注册码作为标识贴在产品的外包装上。除了满足可见(visible)之外,UDI必须满足纯文本形式和自动识别技术(automatic identification and data capture, AIDC). 器械的标签负责人员也必须将每个产品的确实信息发送到“FDA国际特殊医疗器械识别库UDID”内,使公众可以通过访问该数据库查询并下载相关数据(包括从生产,分销到客户使用情况的信息等),但该数据库不会提供器械使用者的信息。
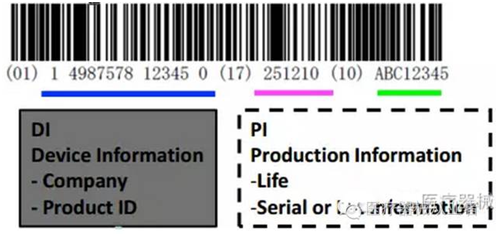
UDI主要是一个由数字或字母组成的编码。由器械识别码(DI)和生产识别码(PI)组成。器械识别码是强制固定的编码,包含了标签管理人员的信息、设备特定版本或型号,而产品识别码不受特别规定,包含了器械生产批号、序列号、生产日期、失效日期以及作为器械管理的生命细胞组织产品的特有识别码。
其次说说GUDID,Global Unique Device IdentificationSystem (GUDID), FDA国际特殊医疗器械识别库。数据库通过AccessGUDID查询系统对外公开。不但可以直接在数据库网页输入标签信息中的UDI的DI码找到产品信息, 还可以通过任一医疗器械的属性搜索(例如仪器识别码Device identifier, 公司或商品名称,通用名称或者器械的型号、版本),但值得注意的是,该数据库不提供器械的PI码。
即UDI定义:唯一器械标识(Unique Device Identification,缩写UDI)是对医疗器械在其整个生命周期赋予的身份标识,是其在产品供应链中的唯一“身份证”。全球采用统一的、标准的UDI有利于提高供应链透明度和运作效率;有利于降低运营成本;有利于实现信息共享与交换;有利于不良事件的监控和问题产品召回,提高医疗服务质量,保障患者安全。
02 UDI的范围和要求? 根据FDA,器械标识包括以下各项,其中,并非所有的类别都要求使用第2项生产识别码: 1、器械识别码: 器械的专利/商标/品牌名称 器械的版本号或型号 器械贴标者(使标签置于器械上、使标签修改、或者使器械引入商业的人) 2、生产识别码: (1)例如批号或批次、序列号、到期日期、生产日期;对于被作为医疗器械监管的人体细胞、组织、或细胞和组织产品,要求使用特定的识别码。除了特定的器械识别码,还需要将有关器械的信息输入到GUDID,其中包括器械识别码,以及下列内容的每一项。生产识别码是不需要的; (2)如果器械使用直接标识,并且直接标识与器械标签上的标识不一致,需要说明哪一个为器械识别码; (3)之前的器械识别码(如果贴标器械为新版本或新型号); (4)器械版本或型号; (5)标签上的生产识别码的类型; (6)FDA市场准入的类型及编号,和列名编号; (7)基于“全球医疗器械命名系统(GMDN)”的产品代码; (8) FDA产品代码; (9)每个包装内的单个器械的数量。 03 FDA授权颁发UDI机构有哪些? FDA目前授权了三大机构GS1, HIBCC以及ICCBBA来负责发行UDI码。根据法规21 CFR 830 subpart C,这些授权机构负责代办FDA授权,审查全美提交的UDI码申请的具体信息,依据规定对申请进行评价。考虑到UDI识别码的出台对于中小型企业的负担,根据法规21 CFR 830 subpart B,以下情况可以豁免或替代UDI标签: 制造和贴标日期早于UDI符合日期的,有3年豁免期 豁免GMP要求的I类医疗器械 被独立包装且单独的,不用于商业流通、使用时包装才被拆除、独立的一次性使用设备 仅用于研究、教学、分析、不用于临床使用的设备 法规ChapterI 812.3 b定义的定制设备 另外,根据UDI FDA官网所称,在UDI 出台的7年内,将逐步完善UDI系统,使其运行地更为有效。对于豁免对象以及申请费用的规定将会进行调整,注册人员应该周期性地随访有关条例。 那不同UDI颁发的机构UDI的形式会有不同么?根据最新的2016年FDA发布的UDI形式说明文件,不同授权机构颁发的UDI会有不同的地方。拿GSI颁发机构举例: 在上表举例中,01到11之间的数字代表的是器械识别码,(11)之后是生产时间,(17)之后是过期时间,(10)之后是器械批号 (21)之后是序列号。 对比HIBCC颁发的UDI形式举例: 在此UDI中,+后面符号代表LIC+ 生产号(Product or catalog number, PCN),/$$后面符号代表过期时间(YYJJJ format)+器械批号,/S 符号后面的字符代表补充序列号,/16D后面的数字代表生产日期。 再对比ICCBBA颁发的UDI形式举例: 需要注意的是,ICCBBA关于血袋的UDI形式有区别于一般医疗器械。更详细的UDI形式解读可参考:FDA 关于UDI式说明文件 虽然各授权机构颁发的UDI有不同的形式,但对于一维条码、二维条码和射频标签(RFID)三种载体都可以认可。为了规范UDI的实施,FDA制定了专门针对UDI的法规: ①21CFR 830---Unique device identification ②21CFR 801 . subpart B---Labeling requirements forunique device identification ③803.32、803.33、803.42、803.52、806.10、820.200、821.25、821.30、822.9. 04 UDI为何如此的重要? FDA在发布UDI法规提案时,确定了几个目标。 1.减少医疗差错; 2.简化将器械使用信息集成到数据系统的操作; 3.更加迅速地识别出现不良事件的医疗器械; 4.更加迅速地为已报告的问题制定解决方案; 5.提供更加迅速、更有效的器械召回解决方案; 6.实现更加重点突出且有效的FDA安全沟通; 7.轻松访问明确器械标识信息的原始来源。 根据UDI最终法规, UDI将会大大减少对美国市场中的医疗器械进行充分识别的障碍。因为UDI可以迅速并明确地识别出器械安全和有效性的关键属性,所以UDI的出现会大大减少此类医疗差错。这是因为这些差错可能是由于器械标识不当或有关其恰当使用的困惑而引起。 UDI系统的优势将会逐步在医疗分配系统中体现出来。各大医疗器械体系在推广UDI的介绍词中都包括了安全性,可追踪性,高效性。该UDI系统能够提高用户的安全、新型医疗器械售后监管和帮助医疗器械的革新。考虑到使用者的安全,当UDI系统完备后,有质量风险的医疗器械将会更容易被追踪和监控,对于不良事件的反馈可以更加有效,审查和分析更加准确,有助于使用者最快时间内解决安全隐患,同时也便于医疗保险的清算。一旦假冒伪劣产品流入市场,查询GUDID数据库关于销售网络的情况,便于使用者判断产品真伪。 UDI系统的出台,另一方面也促进了生产企业的转型,对于产品召回和安全隐患带来的利益损失,督促企业完善质量管理体系,保证产品的有效和安全。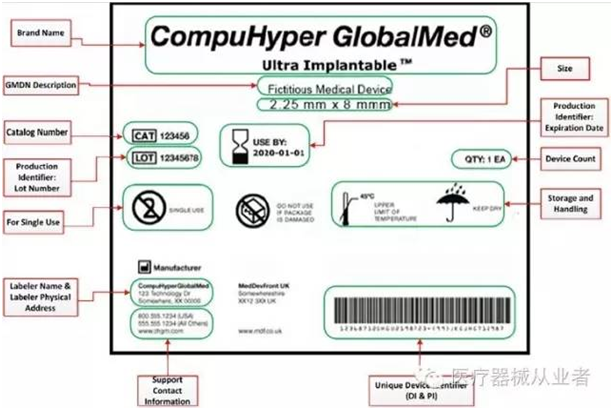
上海市松江区莘砖公路668号双子楼A栋1003室
电话:18964878976 杨浩(销售总监)
展会咨询QQ:515616785
传真:021-31078229
![]()


